Graphene
The TB-model for Graphene¶
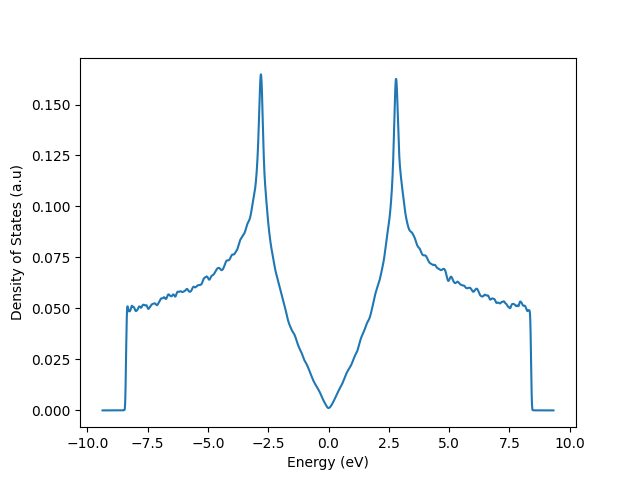
The electronic structure of graphene is well-described by a simple tight-binding model that only uses one \(p_z\) orbital in a hexagonal unit cell with two equivalent carbon atoms. These atoms are located on the different sublattices, A and B, and don't have an on-site energy term.
Although this model is relatively simple, it is used often within the literature. The example below uses graphene to show how to construct a lattice with sublattices and perform a basic calculation using KITE. The script for this example can be found here.
Lattice¶
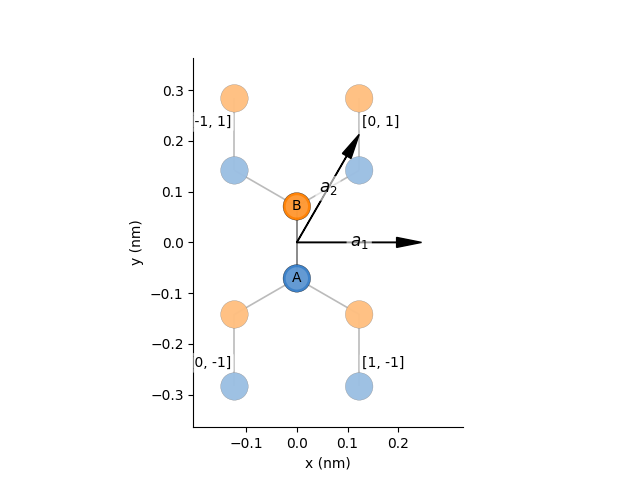
We start by building the pb.lattice for graphene:
- Define the parameter (\(t\) in eV)
- Define the vectors of the unit-cell (\(\vec a_1\) and \(\vec a_2\) in units of \(a\), length of the unit-cell)
- Create a
pb.lattice-object - Define the on-site energies
- Define the hopping parameters
- 1 normal hopping within the unit cell
- 2 rotated hopping \(\pm 2 \pi/3\) to neighbouring cells
- Return the
pb.lattice-object to be used by KITE
We can visualize this lattice using the following code:
KITEx calculation¶
Settings and calculation¶
We can make the kite.Calculation object
kite.config_system as detailed in Sec. Calculation and export the KITE model running
which then creates the necessary HDF file. Next, run the KITEx program and the KITE-tools.
Visualization¶