Tutorial
This tutorial covers the basic aspects that are necessary to start using KITE. You will learn about the different steps in KITE's workflow and how to adjust the different parameters.
The tutorial is structured as follows:
- Learning KITE's workflow
- Building a tight-binding model using
pb.Lattice - How to define the settings for KITE simulations
- How to calculate the different target functions
- Post-processing the results with KITE-tools
- Adding disorder or a magnetic field to the tight-binding model
- How to edit the HDF5-file
- Optimizing the settings for various calculations
First calculation with KITE
Let us perform a simple first calculation with KITE. (Do not worry about the fine details just now, these will be covered later in the tutorial.)
Tip
Run this example from the kite/-folder to have acces to KITE's python package.
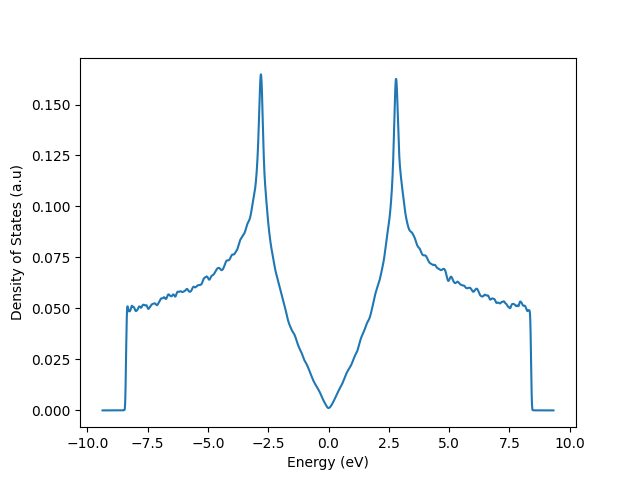
Examples" and "More Examples
Section Examples contains step-by-step examples aimed to expand on key concepts covered by the Tutorial. Additional resources are provided in More examples.